A hemocromatosis foi descrita por primeira vez como unha enfermidade separada en 1889. Non obstante, foi posible establecer con precisión as causas da enfermidade só co desenvolvemento da xenética médica.
Unha clasificación tan tardía foi promovida pola natureza da enfermidade e a súa distribución bastante limitada.
Así, segundo os datos modernos, o 0,33% dos habitantes do mundo corren o risco de desenvolver hemocromatosis. Que causa a enfermidade e cales son os seus síntomas?
Hocromromatosis - que é?
 Esta enfermidade é hereditaria e caracterízase por unha multiplicidade de síntomas e un alto risco de complicacións graves e patoloxías asociadas.
Esta enfermidade é hereditaria e caracterízase por unha multiplicidade de síntomas e un alto risco de complicacións graves e patoloxías asociadas.
Os estudos demostraron que a hemocromatosis é a maioría das veces causada por unha mutación no xene HFE.
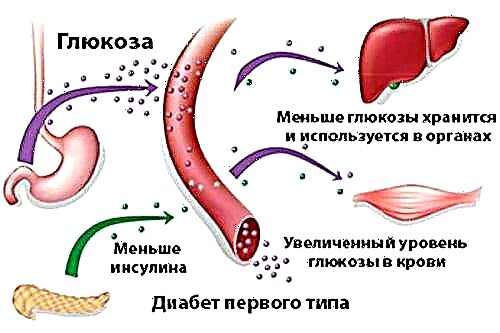
Como resultado dun fracaso xénico, o mecanismo de captación de ferro no duodeno é perturbado.. Isto leva a que o corpo reciba unha mensaxe falsa sobre a falta de ferro no corpo e comece a sintetizar activamente e en exceso cantidades unha proteína especial que une o ferro.
Isto leva a unha excesiva deposición de hemosiderina (pigmento glandular) nos órganos internos. Xunto a un aumento da síntese de proteínas, prodúcese a activación do tracto gastrointestinal, provocando unha absorción excesiva de ferro dos alimentos no intestino.
Así, incluso con nutrición normal, a cantidade de ferro contida no corpo é moitas veces maior do normal. Isto leva á destrución de tecidos de órganos internos, problemas co sistema endocrino e á inmunidade.
Clasificación por tipos, formas e etapas
Na práctica médica divídense os tipos de enfermidade primaria e secundaria. Neste caso, o primario, tamén chamado hereditario, é o resultado dunha predisposición xenética. A hemocromatosis secundaria é consecuencia do desenvolvemento de desviacións no traballo dos sistemas enzimáticos implicados no metabolismo glandular.
Coñécense catro formas de enfermidade hereditaria (xenética):
- clásico
- xuvenil;
- especies hereditarias non asociadas ao HFE;
- dominante autosómico.
O primeiro tipo está asociado á mutación recesiva clásica da sexta rexión de cromosomas. A gran maioría dos casos diagnostícanse este tipo - máis do 95 por cento dos pacientes padecen hemocromatosis clásica.
O tipo de enfermidade xuvenil ocorre como resultado dunha mutación noutro xene, HAMP. Baixo a influencia deste cambio, aumenta significativamente a síntese de hepcidina, un encima responsable da deposición de ferro nos órganos. Normalmente a enfermidade maniféstase na idade de dez a trinta anos.
O tipo non asociado HFE desenvólvese cando falla o xen HJV. Esta patoloxía inclúe o mecanismo de hiperactivación dos receptores de transferrina-2. Como resultado, intensifícase a produción de hepcidina. A diferenza co tipo de enfermidade xuvenil é que no primeiro caso, falla un xene, o que é o responsable directo da produción do encima que une o ferro.
 Mentres que no segundo caso, o corpo crea un estado característico dun exceso de ferro nos alimentos, o que conduce á produción da encima.
Mentres que no segundo caso, o corpo crea un estado característico dun exceso de ferro nos alimentos, o que conduce á produción da encima.
O cuarto tipo de hemocromatosis hereditaria está asociado a un mal funcionamento do xene SLC40A1.
A enfermidade maniféstase na vellez e está asociada a unha síntese inadecuada da proteína ferroportina, encargada de transportar compostos de ferro ás células.
Causas de mutación e factores de risco
 A mutación xenética do tipo hereditario da enfermidade é consecuencia da predisposición dunha persoa.
A mutación xenética do tipo hereditario da enfermidade é consecuencia da predisposición dunha persoa.
Os estudos demostran que a maioría dos pacientes son residentes brancos de América do Norte e Europa, e o maior número de persoas que padecen hemocromatosis observáronse entre os inmigrantes de Irlanda.
Por outra banda, a prevalencia de distintos tipos de mutacións é característica para diferentes partes do globo. Os homes son susceptibles á enfermidade varias veces máis veces que as mulleres. Neste último, os síntomas normalmente desenvólvense despois de cambios hormonais no corpo resultantes da menopausa.
Entre os pacientes rexistrados, as mulleres son entre 7 e 10 veces menos que os homes. As razóns do cambio aínda non están claras. Só a natureza hereditaria da enfermidade está probada irrefutablemente, e tamén se detecta a conexión entre a presenza de hemocromatosis e a fibrose hepática.
 Aínda que o crecemento do tecido conectivo non se pode explicar directamente pola acumulación de ferro no corpo, ata o 70% dos pacientes con hemocromatosis presentaban fibrosis hepática.
Aínda que o crecemento do tecido conectivo non se pode explicar directamente pola acumulación de ferro no corpo, ata o 70% dos pacientes con hemocromatosis presentaban fibrosis hepática.
Ademais, unha predisposición xenética non conduce necesariamente ao desenvolvemento da enfermidade.
Ademais, hai unha forma secundaria de hemocromatosis, que se observa en persoas con xenética inicialmente normal. Os factores de risco inclúen tamén algunhas patoloxías. Así, a steatohepatite transferida (deposición non alcohólica do tecido adiposo), o desenvolvemento de hepatite crónica de diversas etioloxías, así como o bloqueo do páncreas contribúen á manifestación da enfermidade.
Síntomas de hemocromatosis en mulleres e homes
 No pasado, só o desenvolvemento de varias manifestacións sintomáticas graves permitiu diagnosticar esta enfermidade.
No pasado, só o desenvolvemento de varias manifestacións sintomáticas graves permitiu diagnosticar esta enfermidade.
O paciente con excesiva acumulación de ferro sente fatiga crónica, debilidade.
Este síntoma é característico para o 75% dos pacientes con hematochromatosis. A pigmentación da pel aumenta e este proceso non está asociado á produción de melanina. A pel escurece debido á acumulación de compostos de ferro alí. O escurecemento obsérvase en máis do 70% dos pacientes.
O efecto negativo do ferro acumulado sobre as células inmunes leva a un debilitamento da inmunidade. Polo tanto, co transcurso da enfermidade, a susceptibilidade do paciente a infeccións aumenta: desde bastante grave ata banal e inofensivo en condicións normais.
 Aproximadamente a metade dos pacientes padecen patoloxías articulares que se expresan na aparición de dor.
Aproximadamente a metade dos pacientes padecen patoloxías articulares que se expresan na aparición de dor.
Tamén hai un deterioro na súa mobilidade. Este síntoma prodúcese porque un exceso de compostos de ferro cataliza os depósitos de calcio nas articulacións.
Tamén son posibles arritmias e desenvolvemento de insuficiencia cardíaca. Un efecto negativo no páncreas adoita levar á diabetes. O exceso de ferro provoca unha disfunción das glándulas sudoríparas. En casos bastante raros, obsérvanse dores de cabeza.
O desenvolvemento da enfermidade leva á impotencia nos homes. A diminución da función sexual indica signos de envelenamento do corpo con produtos compostos de ferro. Nas mulleres é posible hemorraxias intensas durante a regulación.
 Un síntoma importante é un aumento do fígado, así como unha dor abdominal bastante severa, na aparencia de que non é posible identificar a sistema.
Un síntoma importante é un aumento do fígado, así como unha dor abdominal bastante severa, na aparencia de que non é posible identificar a sistema.
A presenza de varios síntomas suxire a necesidade dun diagnóstico preciso de laboratorio da enfermidade.
Un signo da enfermidade é un alto contido de hemoglobina no sangue, cun contido baixo simultáneo da mesma en glóbulos vermellos. Os indicadores de saturación de transferrina con ferro por baixo do 50% considéranse un signo de laboratorio de hemocromatosis.
Un aumento significativo no fígado cunha alta densidade dos seus tecidos tamén é un signo da enfermidade. Ademais, coa hemocromatosis, obsérvase un cambio na cor do tecido hepático.
Como se manifesta nun neno?
 A hemocromatosis precoz ten unha serie de características: desde as mutacións que a provocaron nas correspondentes rexións cromosomas ata o cadro e manifestacións clínicas características.
A hemocromatosis precoz ten unha serie de características: desde as mutacións que a provocaron nas correspondentes rexións cromosomas ata o cadro e manifestacións clínicas características.
En primeiro lugar, os síntomas da enfermidade a unha idade temperá son polimórficos.
Os nenos caracterízanse polo desenvolvemento de síntomas que indican hipertensión portal. Desenvolve unha violación da asimilación dos alimentos, un aumento simultáneo do bazo e fígado.
Co desenvolvemento da patoloxía, comeza a ascite pesada e resistente aos efectos curativos - a gota que se forma na rexión abdominal. É característico o desenvolvemento de varices do esófago.
Que probas e métodos de diagnóstico axudan a identificar a patoloxía?
 Para identificar a enfermidade úsanse varios métodos de diagnóstico de laboratorio diferentes.
Para identificar a enfermidade úsanse varios métodos de diagnóstico de laboratorio diferentes.
Inicialmente, a mostraxe de sangue faise para estudar o nivel de hemoglobina nos glóbulos vermellos e no plasma.
Tamén se fai unha valoración do metabolismo do ferro.
A proba desferal axuda a confirmar o diagnóstico. Para iso, adminístrase unha inxección dun fármaco glandular e despois de cinco horas tómase unha mostra de ouriña. Ademais, a TC e a RMN dos órganos internos realízanse para determinar os seus cambios patolóxicos - un aumento do tamaño, a pigmentación e un cambio na estrutura do tecido.
A dixitalización xenética molecular permite determinar a presenza dunha parte danada do cromosoma. Este estudo, realizado entre membros da familia do paciente, tamén permite avaliar a posibilidade de que se produza a enfermidade incluso antes da aparición das súas manifestacións clínicas que perturban o paciente.
Principios de tratamento
Os principais métodos de tratamento son a normalización de indicacións de contido de ferro no corpo e a prevención de danos en órganos e sistemas internos. Por desgraza, a medicina moderna non sabe normalizar o aparello xénico.

Limpeza de sangue
Un método común de tratamento é o líquido sanguíneo. Con terapia inicial, elimínanse 500 mg de sangue semanalmente. Tras a normalización do contido en ferro, pasan á terapia de mantemento, cando a mostraxe de sangue se produce cada tres meses.
Tamén se practica administración intravenosa de fármacos ligantes ao ferro. Así, os quelantes permítenlle eliminar o exceso de substancias con ouriños ou feces. Non obstante, un curto período de acción fai necesaria a inxección subcutánea regular de drogas coa axuda de bombas especiais.
Posibles complicacións e prognóstico
Con diagnóstico precoz, a enfermidade pódese controlar de xeito eficaz.A duración ea calidade de vida dos pacientes que reciben atención regular practicamente non difiren da xente de persoas saudables.
Ademais, o tratamento prematuro leva a complicacións graves. Estes inclúen o desenvolvemento de cirrosis e insuficiencia hepática, diabetes, danos nas venas ata o sangrado.
Hai un alto risco de desenvolver cardiomiopatía e cancro de fígado, tamén se observan infeccións intercurrentes.
Vídeos relacionados
Sobre o que é a hemocromatosis e como tratala, no telegrafo "Vive saudable!" con Elena Malysheva: